SPAGHETTI was a program designed to generate simulations of genetic populations with specific segregating molecular markers, incorporating realistic complications such as duplicated loci and segregation distortion (Tinker, 2010). It generated output compatible with standard genetic mapping software and facilitated the testing and demonstrating linkage map construction methods. The software, source code, sample files, and instructions were freely available for public use.
The need for SPAGHETTI arose from the increasing ease of generating genetic linkage maps, which could mask inaccuracies. While modern genomics relied on high-quality maps, even experienced researchers could produce flawed maps. SPAGHETTI addressed this by allowing users to simulate genetic data with known parameters, providing a way to evaluate mapping methodologies objectively.
SPAGHETTI operated via a command line or simple graphical interface, requiring one or two input files. The primary input file specified population parameters (e.g., population type, size, the proportion of segregating loci, missing/mis-scored data, and markers with reversed scoring phase). It supported simulations of F2, doubled haploid, and recombinant inbred populations. A second input file defined the underlying genome, specifying locus names, positions, and other optional parameters. If a user does not provide a genome file, SPAGHETTI could generate a random genome, which could be saved and modified for future use.
The program produced two output files for each simulated population: a “perfect” dataset with all loci scored codominant and without errors and a dataset incorporating the specified abnormalities. These files were output in formats compatible with Mapmaker and JoinMap software. A log file recorded data abnormalities, offering transparency. The simulation also could add random error or “noise” to the process (Tinker & Mather, 1993).
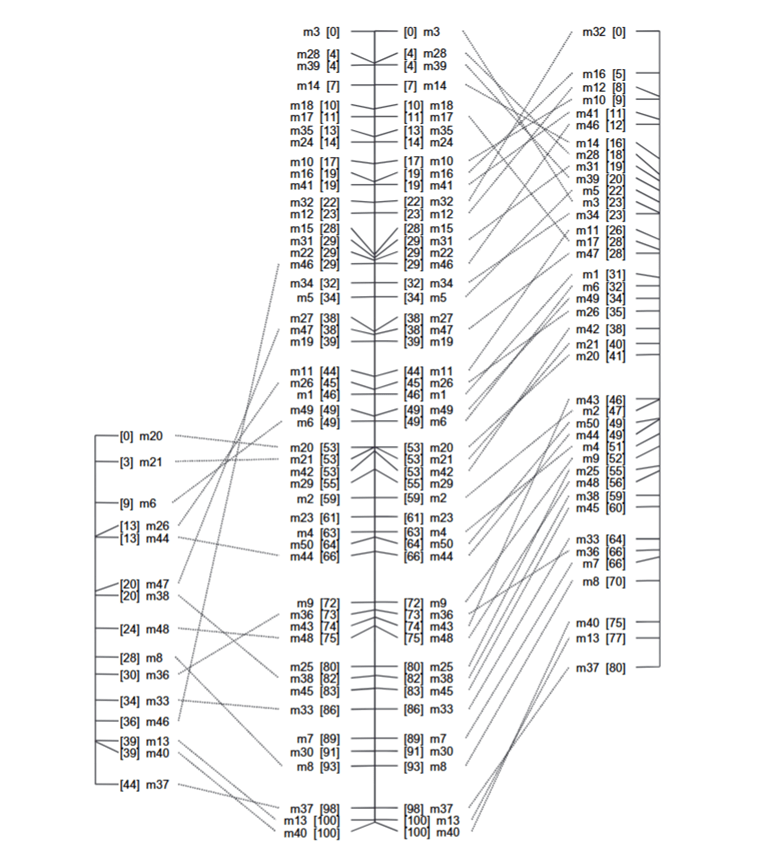
Figure 1: SPAGHETTI simulation (Tinker, 2010).
In this example of use, SPAGHETTI simulated five F5 recombinant inbred populations with a 100 cM chromosome. JoinMap analysis shows that the single population map (left) had fewer markers than the merged map (right). Researchers evaluated the accuracy by comparing the marker positions to the original genome with a mean squared error difference. These results show the benefits of consensus mapping and demonstrate that the original population sizes were too small for aping (Figure 1) (Tinker, 2010).
References
Tinker, N. A. (2010). SPAGHETTI: Simulation software to test genetic mapping programs. Journal of Heredity, 101(2), 257-258. https://doi.org/10.1093/jhered/esp114
Tinker, Nick & Mather, Diane. (1993). GREGOR: Software for Genetic Simulation. Journal of Heredity. 84. 237. 10.1093/oxfordjournals.jhered.a111329.

Leave a comment